Genomic approaches to studying the microbiome
May 18, 2020

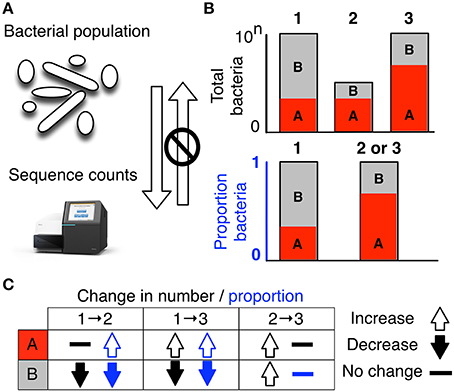
Microbes in a community interact with each other and the host, so it is important to capture as much of the diversity of the microbiome within its phenotypic context as possible.
Understanding how microbial species and the host are related through genome-based and high-throughput techniques is a hot topic in microbiome research.
In my previous post on the plant microbiome I dropped many references to genomics studies which have informed so much of our knowledge on plant–microbe interactions. In this post, I will discuss the modern genomics techniques used in studying the microbiome and the genetic markers which have been set as goal posts.
The main questions when approaching a microbiome study are:
- Who is there? — taxa identification, their abundance and distribution.
- What are they doing? — what functional chemistry is being carried out and what environmental products are being consumed and excreted?
- How are they doing it? — which enzyme pathways are present? Is pathway expression dominant in one species over another?
Abundance = high quantity in a sample.
Prevalence = consistently found across samples.
If both → consistently found in high quantities across samples.
HOW can we study the Microbiome?
Classic microbiology techniques involve isolating and culturing microbes. These culture-dependent techniques are limited — most microbes cannot be grown under lab conditions.
This mismatch between observed cells vs. cultured cells was dubbed the great plate count anomaly.

Culture-independent methods use DNA sequencing:
- 16S rRNA surveys — identify bacteria by sequencing ribosomal RNA.
- Metagenomics — survey all DNA present.
- Metatranscriptomics — measure mRNA expression.
- Proteomics — detect proteins.
- Metabolomics — identify metabolites.
- Metaphenomics — combine multiple omics in context.

Challenges:
- Variation between samples complicates analysis.
- Controls can be difficult (e.g., sterilized soil vs. human longitudinal sampling).
OMICS approaches
The suffix omics = comprehensive, global study (omics.org).

Functional genomics examines how genes and regions across the genome contribute to biological processes.
Questions it can answer:
- Which populations are associated with a phenotype/disease?
- What is the relative abundance of species?
- Which genes are expressed under specific conditions?
- How does a population shift under environmental change?
- What metabolites are produced?
BIOLOGY IS A big data problem
NGS generates vast amounts of sequencing data, requiring computational power and bioinformatics expertise.


The cost of sequencing has plummeted, but data output is growing faster than Moore’s law for computing.
Short reads must be mapped to a reference genome to assemble full genomes.

This is the read alignment problem.

Compared to the human genome (~3 billion bp, 3.2GB), bacterial genomes are tiny (~3–4 million bp, ~3.87MB) (source).
Metagenomics — who is there?
Metagenomics = sequencing all DNA in a community. It identifies taxa present and their functional genes.

Metagenomics ≠ meta-analysis. Metagenomics = genetic survey of an environment. Meta-analysis = comparison across datasets.
16S ribosomal RNA
16S rRNA is the barcode gene in microbial ecology. It’s:
- present in all bacteria
- highly conserved within species
- has variable regions useful for taxonomy
- sometimes insufficient (e.g., E. coli vs. Shigella >99% identical Devanga Ragupathi et al., 2017)

Carl Woese pioneered 16S phylogenetics in the 1970s, proposing the 3-domain system (Bacteria, Archaea, Eukarya) Woese et al., 1990.

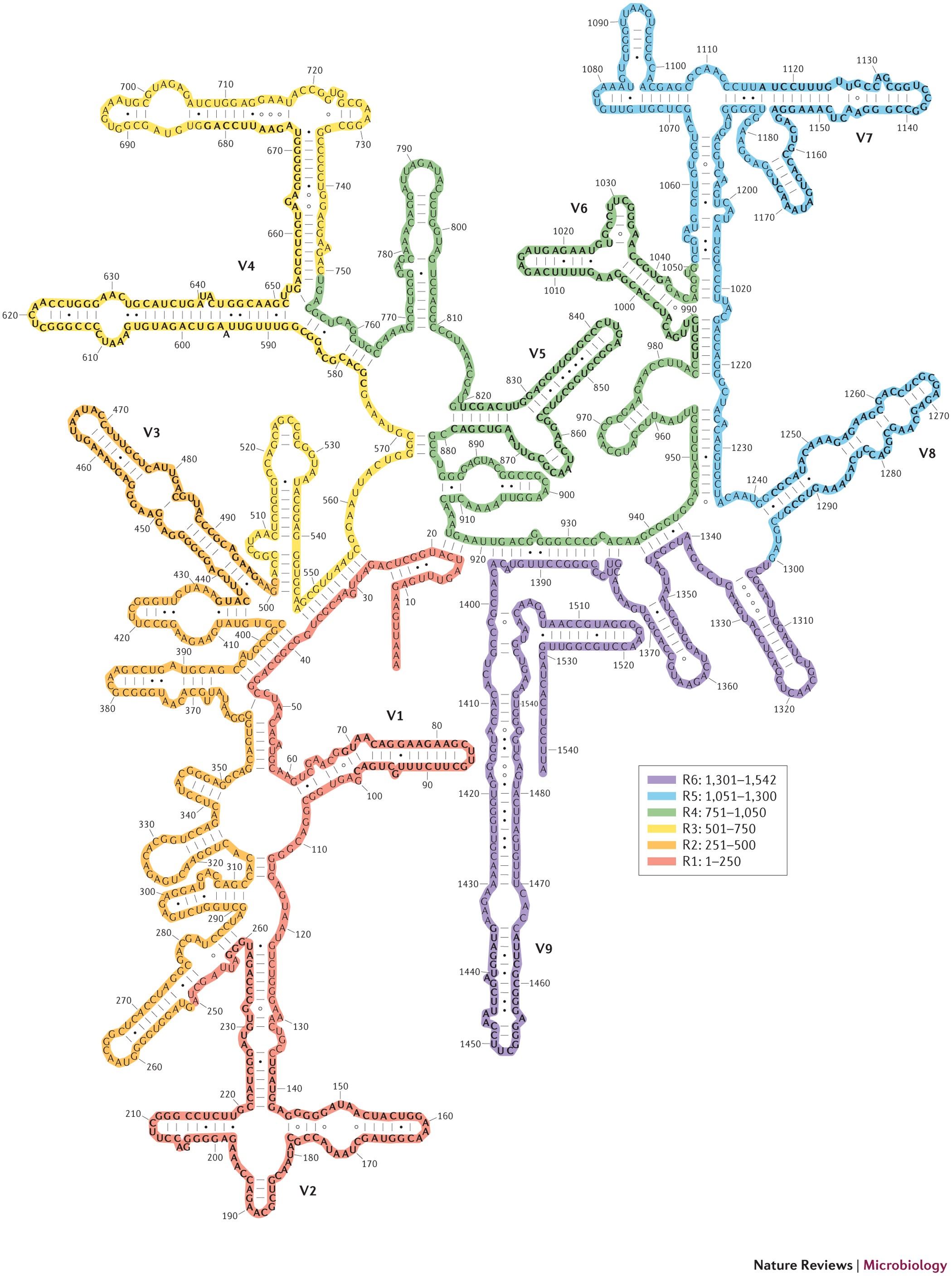
Advantages of 16S:
- cheap
- high sample throughput
- huge databases
Disadvantages:
- not quantitative (multiple 16S copies per genome)
- limited resolution (often genus-level)
- compositional (relative, not absolute)
Metagenomic whole genome sequencing
WGS captures all DNA, including viruses/fungi missed by 16S.

Steps:
- Extract DNA
- Prepare library + sequence
- Map reads to marker genes, pangenomes, or protein DBs (MetaPhlAn, HUMAnN2)
- Or assemble de novo (de Bruijn graphs)


Output: complete genomes (rare) or contigs (common).
Metatranscriptomics — what can they do?
Looks at RNA transcripts to see which genes are actively expressed.
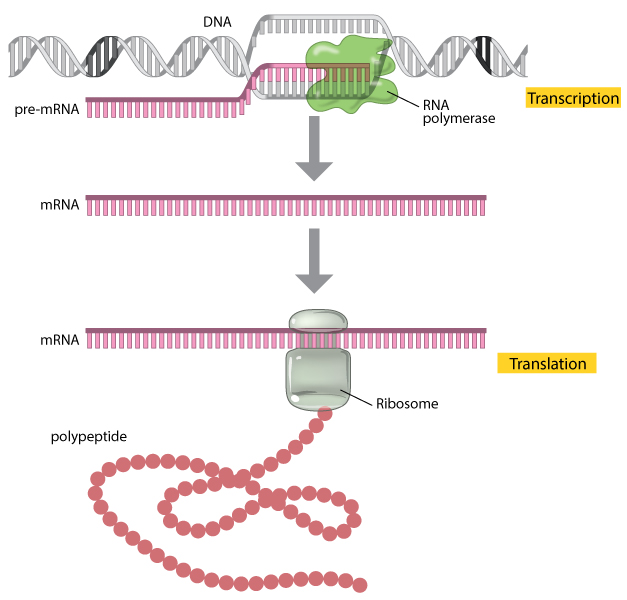
mRNA = single-stranded transcript coding for proteins. By sequencing RNA (RNA-Seq), we capture active functions.
Process:
- Extract RNA
- Reverse transcribe to cDNA
- Fragment + sequence
- Align reads → quantify expression

Approaches:
- Reference-based alignment (STAR, HiSAT2, Bowtie)
- Splice-aware alignment (TopHat2, etc.)
- Pseudo-alignment (kallisto Bray et al. 2015)
Challenges:
- bacteria lack polyA tails → high rRNA contamination
- many microbes lack reference genomes

Differential gene expression analysis
Heatmaps, PCA, clustering are used to visualize expression.

Proteome
The proteome = all proteins expressed. More accurate than transcriptome since not all transcripts → proteins.
Metabolome
Metabolites = intermediates + end products of metabolism. Reveal biochemical pathways.
Metaphenome
The metaphenome = combined functional output of a microbial community in its environment.

Attempts to connect in situ expression to ecosystem-level functions like cellulose decomposition, methanogenesis, sulfate reduction.
Conclusion
I hope this foray into microbiome genomics illuminated some of the methods shaping modern research. In future posts I’ll dive deeper into workflows using real NGS data.