The CrAssphage story
Mar 30, 2021
Metagenomics and Discovering Viruses from Sequences
Analyzing shotgun metagenomics data has become an exciting method of virus discovery in modern microbiology. Research insights gleaned this way are deemed in silico - or by computer.
Most of the virus genomes we currently have access to come from metagenomics (all DNA sequences from multiple species found in a single sampled environment). Even though a particular virus can be highly abundant in its environment, more often than not its host has not been identified and therefore has never been cultured in the lab.
From these sequences we know that the human gut virome is abundant in bacteriophage. Gut virome compositions are also highly unique and stable to each individual.
Up to 80% of all viral sequences in a shotgun metagenomics sequence have no known homology to a reference database. These unknown viral sequences have been termed viral dark matter and present a unique opportunity to mine new viral signals and discover new viruses from publicly available genomics data.
One example of a virus discovered through bioinformatics and metagenomics is crAssphage (cross-assembly phage), named after the assembly software that aided in its discovery.
CrAssphage Fun Facts
-
CrAssphage is the most abundant phage in our gut.
-
It can account for up to 90% of the reads in the viral portion of the gut metagenome and around 22% of the reads of the total metagenome in some individuals.
-
98–100% of healthy adults from Western samples carry at least one type of crAssphage.
-
CrAssphage-like sequences have been found in faecal samples from remote regions such as rural Malawi and the Amazonas of Venezuela.
-
It is found in all age groups (from as young as 1 year old to 65+) and is thought to be acquired during early childhood.
-
It is a benign gut inhabitant as no associations between it and diet or health have emerged.
-
CrAss is “a cosmopolitan virus that may have coevolved with the human lineage and is an integral part of the normal human gut virome.”
-
Found in human populations all over the world and crAssphage populations are very region-specific (you can identify the region someone is from by their crAssphage population!).
Cross-Assembly (cr-Ass)
So what is cross-assembly?
Cross assembly involves combining reads from multiple metagenomics datasets and making a single assembly from these reads - i.e., a single assembly from two or more metagenomes. The resulting contigs may be interpreted as metagenomic entities or shared traits between sampled environments.
This makes CA a reference-independent comparative metagenomics method whereby you compare all reads of multiple different samples. If a sequence or number of reads is common among all samples (a cross-contig), or among some and not others, you can deduce interrelationships between their metagenomes.
Cross-contigs are contigs that are common across two or more metagenomes.
This is how researchers discovered CrAssphage - they found a sequence homologous across all subjects and then investigated it further.
Benefits of Cross-Assembly
When analyzing metagenomics data, the first step is often aligning reads to a reference genome. In viral studies, there are no comprehensive reference genomes, and databases are limited. If your goal is viral discovery, you must uncover patterns independently - this is where deep learning and alternative methods such as cross-assembly become powerful.
Discovery of CrAssphage Approach (Dutilh, 2014)
Data:
Re-analysis of publicly available viral metagenomes from faecal samples of 12 individuals (4 pairs of monozygotic twins and their mothers; families unrelated).
Software: Used the cross-assembly program CrAss.
Data Analysis Approach:
- 1,584,658 metagenomic reads
- Cross-assembly produced 7,584 cross-contigs
- One contig, contig07548, contained reads from all 12 subjects - suggesting a ubiquitous viral entity
- Depth profile binning and homology binning identified contigs from the same genome
- Separate assembly yielded a ~97 kb circular crAssphage genome
Predicting the Host
Since phages require hosts, the authors used co-occurrence profiling to identify correlations between crAssphage reads and bacterial reads.
CRISPR Spacer Prediction
CRISPR spacers are fragments of viral DNA incorporated into bacterial genomes as immune memory. By comparing crAssphage sequences to CRISPR spacers from 404 intestinal bacterial strains, they predicted the host belonged to the phylum Bacteroidetes.
Since Bacteroidetes are highly abundant in the gut, this made ecological sense.
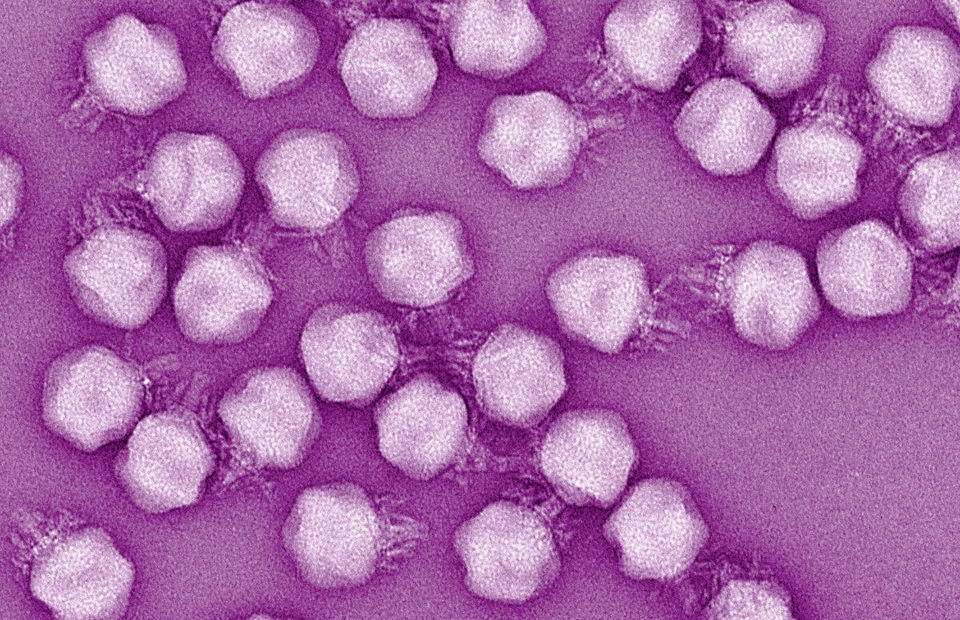
First Isolated CrAssphage: ΦcrAss001
The first crAss-like representative was successfully cultured by Andrey Shkoporov and named ΦcrAss001.
Findings:
-
Circular genome, 102 kb
-
Podovirus-like morphology confirmed by electron microscopy
-
Host: Bacteroides intestinalis
-
Replicates without disrupting host proliferation despite lacking obvious lysogeny genes

Isolation Approach
-
Faecal samples from 20 healthy Irish adults enriched for phage
-
Samples pooled and used to infect 54 gut bacterial strains
-
After enrichment and sequencing, a 102.7 kb contig dominated reads in Bacteroides intestinalis
-
Genome comparative analysis confirmed it was a crassphage homolog

Future CrAssphage Study
Further research into the function and ecological role of this ubiquitous gut virus will be fascinating. Given its abundance and long co-evolution with humans, it likely occupies an important niche in the gut microbiome.
Sources
-
Reference-independent comparative metagenomics using cross-assembly: crAss
-
A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes
-
Global phylogeography and ancient evolution of the widespread human gut virus crAssphage
-
Reference-independent comparative metagenomics using cross-assembly: crAss
-
Streamlining CRISPR spacer-based bacterial host predictions to decipher the viral dark matter